1-D electron density profiles
This TUI guides the user towards the objective indicated in the title by offering a sequence of menus, each of which is displayed individually on the screen. In each menu the user must choose the most appropriate option, which in turn can request additional input.
Three different flowcharts show how to obtain three different types of electron density profile along a given direction.
PAMoC-TUI isn't a visualization program, so that it doesn't provide any
graphical output directly. Its results are printed in tabular form on the
output file and saved on a CUBE file.[L1CUBE
File Format
(PAMoC Manual)] Eventually, the TUI can launch a
suitable visualization program which is able to read the CUBE file.[L1CUBE File Format
(PAMoC
Manual)]
- Radial electron density profile
- Planar electron density profile
- Volumetric or position electron density profile
- Output of 1-D electron density profiles
- References
- Links
1. - Radial electron density profile
The radial electron density at a given radius is the number of electrons in a infinitesimally thin spherical shell at that radius. Its units are e⋅bohr−1.
To obtain the profile of this density along the radius we'll proceed as shown below in Flowchart 1:
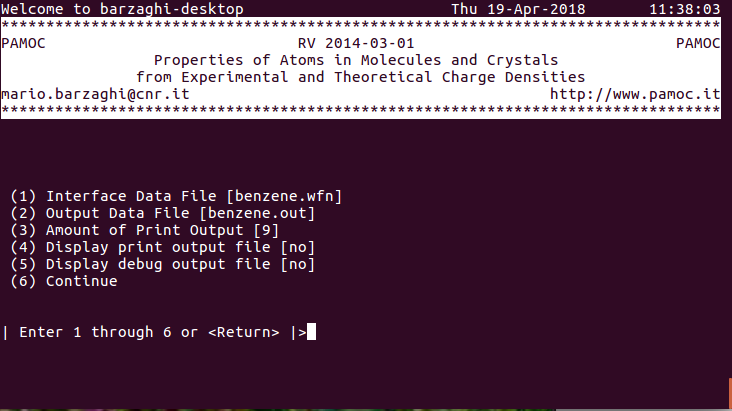 |
(1) (subroutine Screen000) choose item 6 ⇓ |
|
(2) (subroutine Screen000) choose item 5 ⇓ |
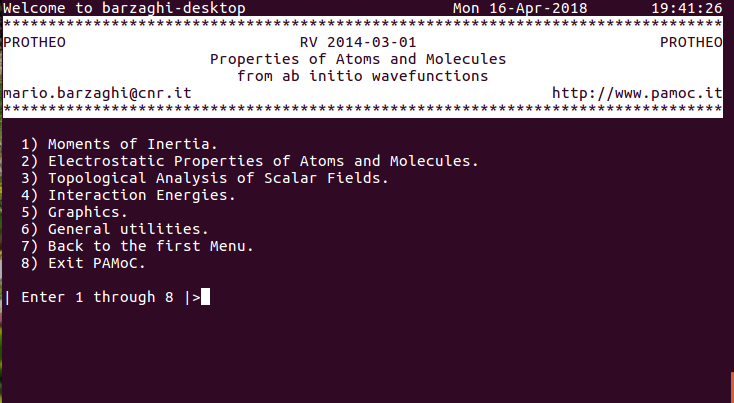 |
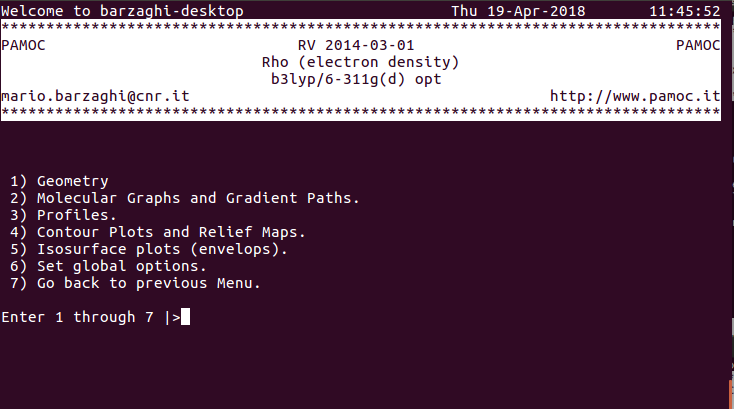 |
(3) (subroutine Screen500) choose item 3 ⇓ |
|
(4) (subroutine Screen530) choose item 2 ⇓ |
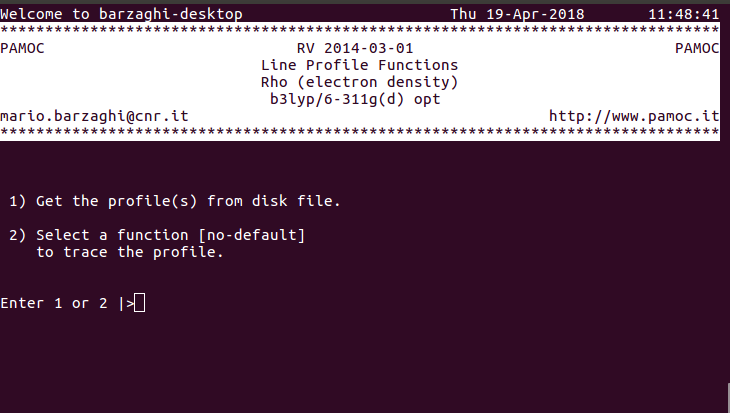 |
 |
(5) (subroutine Screen134) choose item 4 ⇓ |
|
(6) (subroutine Screen530) choose item 2 ⇓ |
 |
 |
(7) (subroutine Screen530) choose item 2 ⇓ |
|
(8) (subroutine Screen530) enter the cartesian coordinates of the center of the spherical shell ⇓ |
 |
 |
It is clear from the asympotic value of the electron count function G(r) in Figure 1, as well as from the numerical results reported in the print-output file (see Section 4), that the presence of cusps at the nuclear centres destroys the accuracy of the numerical procedure used. In the example of Figure 1, the number of electrons in benzene is overestimated by 4.31 e (about 10%).
The error is even higher if the center of the spherical shell is located at a nuclear centre (screen 7, option 1, Flowchart 1), like a carbon atom (Figure 2a, error = 15.41 e or 36.7%) or an hydrogen atom (Figure 2b, error = 54.42 e or 130%).
 |
From the examples above (Figures 1 and 2), it appears that the radial electron density of a molecular system is strongly dependent on the origin of the spherical shell, and can be of some interest only if the origin is placed in some special point, like a center of symmetry (Figure 2). On the other hand, it is particularly suited to describe the electron density of an isolated atom.
2. - Planar electron density profile.
The planar electron density at a given point on a line is the number of electrons in a plane perpendicular to that line in that point. Its units are e⋅bohr−1.
To obtain the profile of this density along the line we'll follow the steps outlined in Flowchart 1 up to the screen number 6, where we will choose the option number 3 and proceed as shown below in Flowchart 2:
|
(6) (subroutine Screen530) choose item 3 ⇓ |
|
 |
(7) (subroutine Screen530) choose item 3 ⇓ |
 |
On the other hand, choosing option number 1 or 2 on screen number 7 in Flowchart 2, yields the following graph
 |
The error in the electron count is still influenced by the presence of cusps on nuclear centres, but to a lesser extent than the integration on the unit sphere. In fact, it is underestimated by 0.5 electrons (1.20%) in the case of Figure 3, and by 3.4 electrons (8.12%) in the case of Figure 4, respectively.
3. - Position electron density profile.
The position electron density at a given point in 3-D space is the number of electrons contained in an infinitesimally small volume at that point. Its units are e⋅bohr−3.
The electron density values at a number of points along a line in 3-D space provide an electron density profile along that line. It can be obtained by selecting the option number 1 on the screen number 5 in Flowchart 1, and going through the steps shown in Flowchart 3:
|
|
(5) (subroutine Screen134) choose item 1 ⇓ |
|
(6) (subroutine Screen134) choose item 1 ⇓ |
 |
 |
(7) (subroutine Screen3) choose item 1 ⇓ |
|
(8) (subroutine Screen2) at the prompt enter “atom 10” for P0 ⇓ |
 |
 |
(9) (subroutine Screen2) confirm striking “RETURN” ⇓ |
|
(10) (subroutine Screen2) at the prompt enter “atom 7” for P1 ⇓ |
 |
 |
(11) (subroutine Screen2) confirm striking “RETURN” ⇓ |
|
(12) (subroutine Screen3) choose item 1 ⇓ |
 |
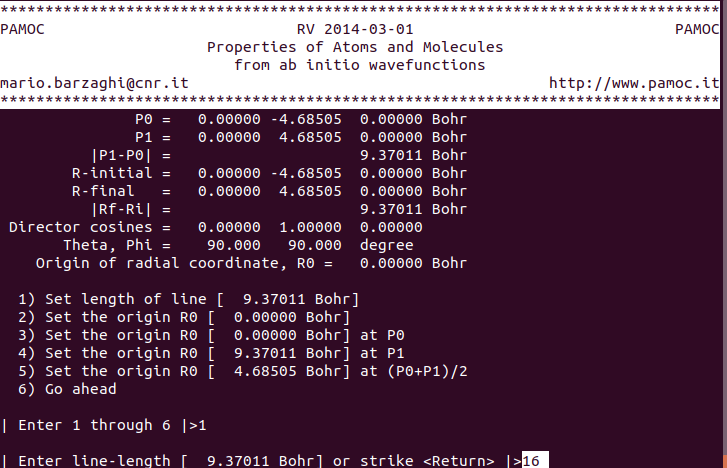 |
(13) (subroutine Screen3) enter “16” for line-length (bohr) ⇓ |
|
(14) (subroutine Screen3) choose item 5 ⇓ |
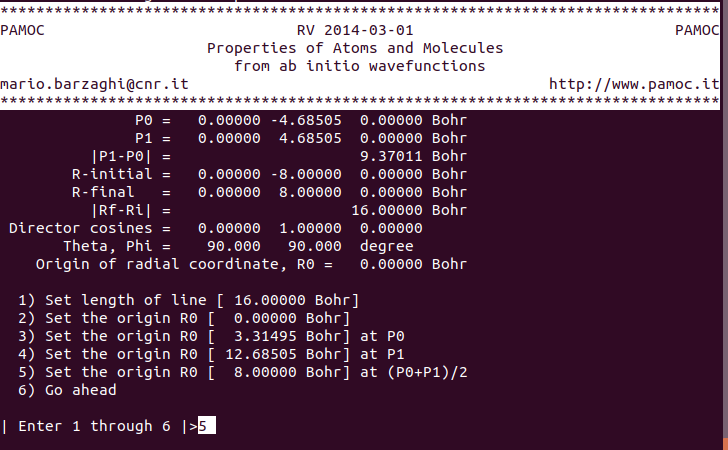 |
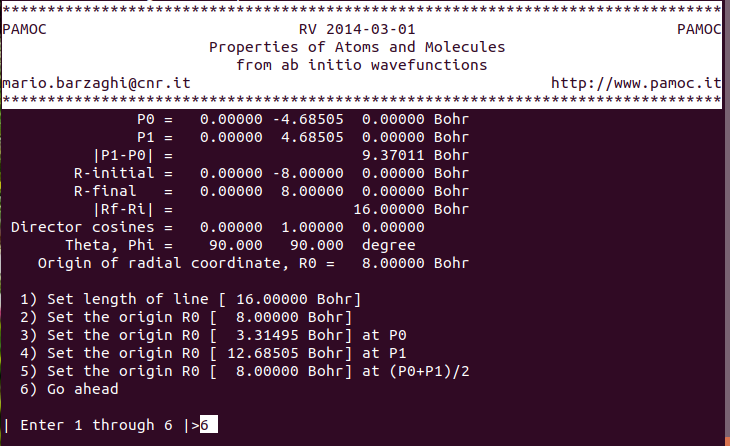 |
(15) (subroutine Screen3) choose item 6 ⇓ |
 |
The profiles in Figures 3 and 5 have some analogies as well as significant differences. Both figures describe the trend of the electron density along a given direction, but the values shown in Figure 5 refer to the electron density at each point of the space along that direction, while those shown in Figure 3 give the value of the total electron density in the plane perpendicular to the given direction.
4. - Output of 1-D electron density profiles
The graphs of Figures 1-5 are not produced directly by the PAMoC-TUI.
Instead the latter provides numerical results in the form of a block data,
which is part of the print-output file. The block data (shown in the box below)
can be extracted from the print-output file and then read by
GRACE.[L2"Grace (plotting tool)",
Wikipedia,
The Free Encyclopedia, L3GRACE website], a free 2D graph plotting tool,
for visualization. All Figures 1-5 have been obtained using GRACE. The block
data reported in the box below was used to generate Figure 1.
Using GRACE allows the user to change visualization of data as desired.
However, as indicated by the last line in the box extracted from the
print-output file (see above), the PAMoC-TUI also generates a
CUBE file,[L1CUBE File Format
(PAMoC Manual)] which
contains the 1-D electron density profile. The default name of the
CUBE file, is pamoc.g1d,
but the user is allowed to change it by entering a new filename at the
prompt in the following screen:
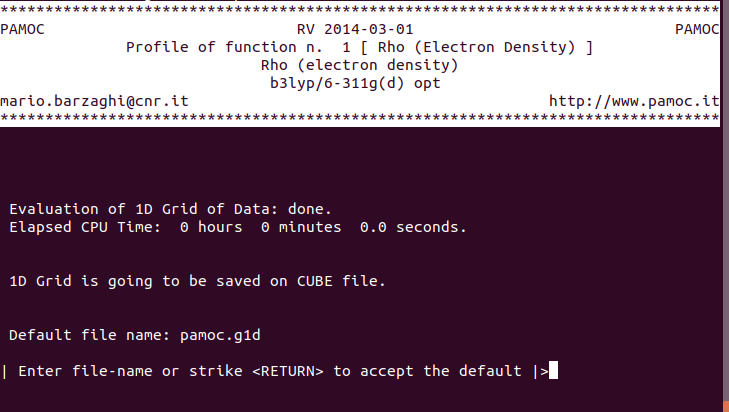 |
(subroutines Screen530 and get_file) |
which is always presented as soon as calculation of the 1-D electron density profile is done.
References
Links
- “PAMoC User's Manual: CUBE File Format”. Online
resource:
https://www.pamoc.it/tpc_cube_file_format.html.
Accessed ?, 2018.

- Wikipedia contributors, "Grace (plotting tool)", Wikipedia,
The Free Encyclopedia, https://en.wikipedia.org/wiki/Grace_(plotting_tool)
(accessed May 1, 2018).
- “GRACE: GRaphing, Advanced Computation and Exploration of
data”. Online resource:
http://plasma-gate.weizmann.ac.il/Grace/.
Accessed May 1, 2018.