This keyword specifies the function which has to be used to
dampen atom-atom dispersion energies.
Available functions are reported in the following table:
Damping Functions (the default
choice is highlighted)
The need for a damping function arises from the fact that
the dispersion energy behaves as r-6 and becomes physically
unrealistic at small distances r where it diverges.
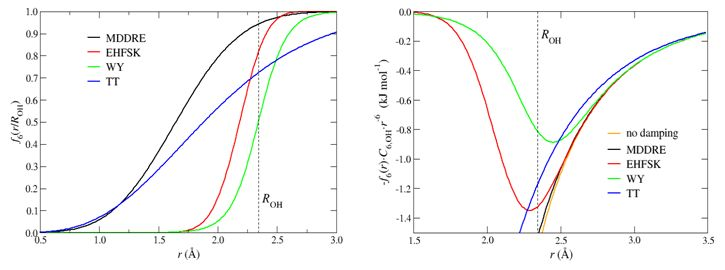
The trend of the four damping functions
(MDDRE,
EHFSK,
WY, and
TT) along the distance r is shown
in the left figure below, whereas their effects on the
C6r-6
term for the OH interaction potential is shown in the right figure.
It appears that the damping strength by
EHFSK function is between those by
funtions MDDRE and
WY, with
WY function being the strongest.
Related Keywords
(AtmParDsp |
apde | adp)
specifies which set of dispersion atomic parameters has to be used for
calculation of atom-atom dispersion interaction energies.
Related Topics
Energies of nonbonded interactions
References
"Transferable ab initio intermolecular potentials. 1. Derivation
from methanol dimer and trimer calculations"
W.T.M. Mooij, F.B. van Duijneveldt, J.G.C.M. van Duijneveldt-van de Rijdt,
B.P. van Eijck
J. Phys. Chem. A1999, 103, 9872-9882.
"Empirical correction to density functional theory for van der Waals
interactions"
Q. Wu, W. Yang
J. Chem. Phys.2002, 116, 515-524.
"Hydrogen bonding and stacking interactions of nucleic acid base
pairs: A density-functional-theory based treatment"
M. Elstner, P. Hobza, T. Frauenheim, S. Suhai, E. Kaxiras
J. Chem. Phys.2001, 114, 5149-5155.
"Accurate Description of van der Waals Complexes by Density
Functional Theory Including Empirical Corrections"
S. Grimme
J. Comput. Chem.2004, 25, 1463-1473.
"Semiempirical GGA-Type Density Functional Constructed with a
Long-Range Dispersion Correction"
S. Grimme
J. Comput. Chem.2006, 27, 1787-1799.
"Density Functional Theory Augmented with an Empirical Dispersion
Term. Interaction Energies and Geometries of 80 Noncovalent Complexes
Compared with Ab Initio Quantum Mechanics Calculations"
P. Jurecka, J. Cerny, P. Hobza, D. R. Salahub
J. Comput. Chem.2007, 28, 555-569.
"An improved simple model for the van der Waals potential based
on universal damping functions for the dispersion coefficients."
K. T. Tang, J. P. Toennies
J. Chem. Phys.1984, 80, 3726-3741.
"A Universal Damping Function for Empirical Dispersion Correction
on Density Functional Theory"
Yi Liu, W. A. Goddard III
Materials Transactions2009, 50, 1664-1670.